AlphaFold 3: what's new in techbio
May 8th was my birthday, and to celebrate it, Alphabet decided to release the AlphaFold 3 paper (thank you, Demis, John and Max, I know you timed it for me!). Those who know me are aware of my special relationship with this software brand. After AlphaFold 2 made my PhD obsolete overnight, I wrote two blog posts on it, first right after CASP14, and then when the paper was published, which led to significant Twitter movement and had me featured in a bunch of international newspapers like Le Monde, Forbes or El Mundo. Every time I go to a conference, someone reaches out to tell me how much they loved my post, or how they got interested in AI for biology thanks to it. The clout works, of course. So, I decided to leave my birthday candles lit for a little longer than needed, and start writing this blog post.
If you do not want to read any more and just get a glance of what has changed, read no further:
- The two main real world outcomes are significant improvements in the prediction of
- protein-ligand complexes (about 30-40% better than AutoDock Vina, a tool which at 15 years of age remains quite high in benchmarks), and
- protein-DNA complexes (>2x better than Baker’s lab’s RoseTTAFoldNA, which was the state-of-the-art).
- The paper also reports a significant improvement on antibody-antigen complex prediction, which depending on your camp may mean a lot or absolutely nothing.
- Yet, in stark contrast with AlphaFold 2, there is no publicly available code, and no announcements of whether it will be made available in any form (commercially or otherwise). The authors have published an AlphaFold 3 server, but it is limited to 20 predictions a day and a very reduced set of ligands.
- While the validation of the method is significantly weaker that in the previous generation, the paper is undeniably a major scientific breakthrough, but its translation into the clinic, or even industry at large, is still an open question.
Since I have now fulfilled the purpose of telling you what happened, and because, what the heck, I started this on my birthday, I am going to write this blog post in a slightly non-traditional path. First, I am going to do a slight dive into the technical details of the model architecture, and speculate on what is important and what it is not. Then, I will discuss how it has impacted different areas of techbio – or rather, what they claim that they have done, and what I think they are allowed to claim based on the paper. Then, I am going to echo many colleagues and complain starkly about the way that this work was published, and finish with a some speculation on the next steps from here.
What has changed: the technical dive
If you download the Nature paper, you might be scratching your head, wondering if you have downloaded the AlphaFold 2 paper instead. It is not just that they used the same graphic designer: the overall architecture looks eerily similar.
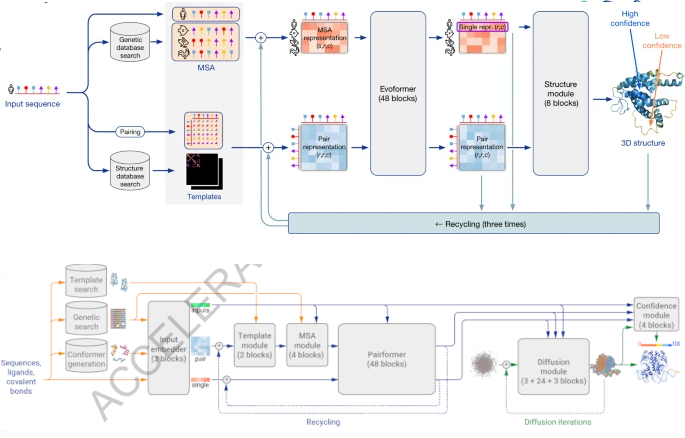
A comparison of the AlphaFold 2 (top) and the AlphaFold 3 (bottom) architectures. The graphic designer is similar, and so is the blueprint.
A cursory glance at the first few Figures will show that the big picture is approximately similar from one version to the next. The model is divided into two conceptual blocks: one that captures coevolutionary information from a multiple sequence alignment, and another one that transforms these inferences into the three-dimensional coordinates of a protein. Added to this is, as well, a confidence prediction that reports when a model is trustworthy. If AlphaFold 2 represented a paradigm shift, AlphaFold 3 appears to share the same blueprint.
One of the first surprises is that the model’s emphasis on coevolutionary information has been significantly reduced. Let me clarify this. I tend to argue that the ability to extract information from the multiple sequence alignment is really the secret sauce in AlphaFold 2. Coevolution contains a lot of information about the structure of the protein (see the box below for more details), and the algorithm in AlphaFold 2 was a very clever way to extract it. We know that that a protein has too many possible conformations to just explore them manually and find the best one, even with a clever search algorithm. The approach of mining pairs of residues that were structurally close, and then building a reasonable model by constrained optimization had been established well before AlphaFold 2.
To understand the role of coevolution in protein (or biomolecule!) structure prediction, I would like you to imagine a protein vital for life. The structure of this protein relies on many amino acids being in contact, and establishing intermolecular interactions that are favourable for its fold. Imagine, for example, that in this protein there is a negatively charged amino acid, like glutamate, that is positioned close to a positively charged amino acid, such as lysine. Although these two amino acids are quite distant in the amino acid sequence, they are close in space because proteins are three-dimensional objects. Therefore, their opposite charges can interact, attracting themselves and keeping the protein’s scaffold in place.
Imagine now that one of these two amino acids, perhaps the glutamate, is mutated by a random accident of evolution. Since this protein is essential to life, the disruption of the protein structure by the destabilising mutation will result in cell death. Therefore, this mutation will not be propagated. We will not encounter sequences with this mutation in any living organisms – unless the other amino acid also experiences a mutation that counteracts the first one (for example, if the glutamate mutates to a positive amino acid, the lysine might mutate to a negative one). We say that there is an evolutionary pressure induced by the structural proximity of the amino acids – and with clever statistical methods we can track these down.
The strategy used in the previous generation of AlphaFold was to combine two learned representations, a “pair representation”, capturing inferred structural information, and a “MSA representation”, capturing coevolution, with the two representations continuously exchanging information to reach an inference. I like to imagine this as an iterative process: the MSA representation is used to identify correlations that point to residues interacting. Then, once an interaction has been established, its environment is re-explored to find further information. In this version, however, the two representations are merged into a single pair representation, and a somewhat downscaled network is used to process it.
The second surprise is that the structure module is, well, no longer the structure module. If you recall the original architecture, there were two interesting parts in the structure module. First, the protein was represented as a point cloud: every residue was a point in space, centered in the backbone. The local structure around the alpha carbon is assumed rigid – which is a fairly good approximation! Second, the model used a clever piece of machinery, invariant point attention, which used transformer-like attention while enforcing that the model respected the physical symmetries. When you rotate a physical object, some properties (for example, its weight) remain invariant, whereas others (for example, its velocity) maintain their magnitude but change their direction alongside the rotation. This inductive bias makes the model learn physical information more rapidly.
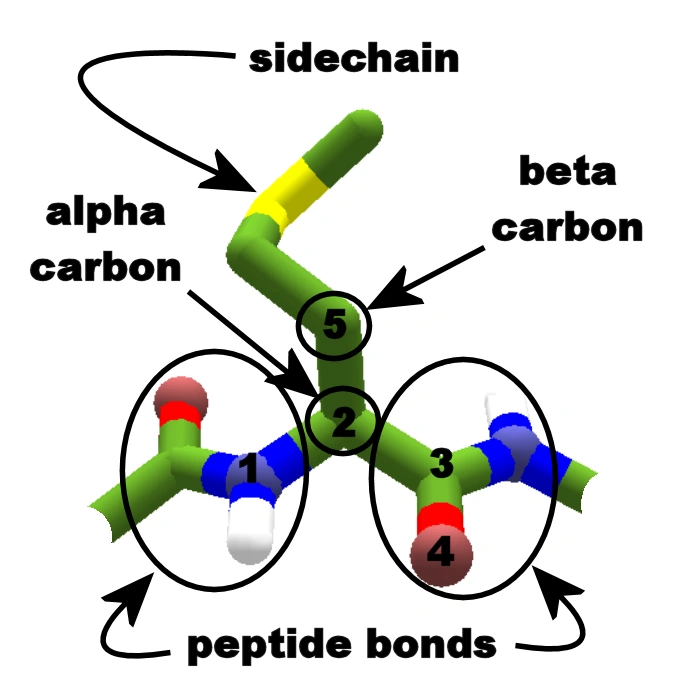
All amino acids share a common backbone, consisting of a central alpha carbon, a nitrogen atom and a carbon atom. Nineteen out of the twenty proteinogenic aminoacids also have a side chain, which starts with a beta carbon and depends on the identity of the amino acid (for example, in alanine, it is just the beta carbon with its corresponding hydrogen atoms). In AlphaFold 2, the amino acids were defined by their backbone, which was assumed to be rigid and ideal, and represented only by the position of the alpha carbon and its relative rotation in space. The side chains were defined by torsion angles, predicted by a different part of the network. Image reproduced from the Foldit wiki.
In AlphaFold 3, however, they drop this completely. The architecture is not even attention-based anymore, but instead uses a “relatively standard” diffusion model, the same kind of technology that is behind DALL-E, StableDiffusion and others. The positions of the atoms are first set at random, then iteratively updated by a “denoising” neural network until they reach the final prediction. Notably, every atom in the protein – backbone atoms as well as side chains – is free to move in space, making away with the “residue gas” approach.
Oh, and talking about things that AlphaFold 2 made popular that AlphaFold 3 decided to drop, there is equivariance. I personally find this hilarious. In the aftermath of CASP14, SE(3)-equivariance was what everyone was talking about – I remember how Fabian Fuchs, who completely independently had developed an SE(3)-equivariant transformer was invited to give a talk on his work at pretty much every techbio event I could think of. On one week, I swear I listened to his (great) talk at least three times. The word SE(3)-equivariance has pretty much been accepted as a sign of quality, so much so that accomplished deep learned Thomas Kipf – who pretty much kickstarted graph neural networks – sparked a huge controversy last year after he suggested equivariance wasn’t really that important. He did have his “I told you so” moment. I tip my hat to you, Thomas.
Anecdotes aside, I find it fantastic that they have managed to achieve so much with the diffusion model, given that most of the reports of atomic diffusion I have heard have failed more or less catastrophically. I am particularly surprised by their performance on ligands, especially considering that they have used the PDB as main source of data. They do pull a bunch of tricks to train the model on almost every other variety. For protein complexes, the run a cross-distillation approach where they predict multiple complexes with AlphaFold Multimer v2.3, and train on that (this is especially helpful to reduce hallucinations). For RNA they use predicted structures, and for DNA they use a bunch of augmentations like data from JASPAR. For ligands, however, they use nothing but the PDB.
I am so surprised by AlphaFold 3’s performance on ligands that I have even started wondering if the model has in some way learned the physics of intermolecular interactions. Some predictions using the server have shown that it roughly reproduces the structure of electrolytic solutions and that it forms a pseudo-membrane when one adds a bunch of lipids to a transmembrane protein, suggesting that it has a measure of physicochemical understanding. A recent benchmark using the provided server suggests that the ranking score also captures important features of how mutations change protein-protein binding affinity. However, protein-ligand interaction is one of the problems in the frontier of biophysics, and I just cannot believe that AlphaFold 3 has learned everything it needs from the very limited data available on the PDB. If they had used some augmentations with plentiful data – say, to include a few DEL screens in a similar way to their transcription factor data – I could doubt, but right now this seems a wild guess rather than a measured scientific statement.
It is perhaps also ironic that AlphaFold3 is using such a simple diffusion strategy, when most other protein modelling approaches have relied on the residue gas, as some have noted. Diffusion models have been used extensively in protein design work, with some of the most popular methods, like Chroma and RFDiffusion-AA all use idealised residues defined by their position and rotation. They also employ a number of other tricks e.g. bond embeddings and special loss functions. The outstanding performance of AlphaFold 3 with such a simple architecture will probably have the entire field thinking – look out for some exciting improvements on diffusion-based design in the next year.
The final detail that the authors have provided the training curve of the model, showing how performance changes on a variety of tasks. It is interesting that learning occurs rapidly, saturating at about 10% of the training – this is reminiscent of what Mohammed AlQuraishi and the rest of the OpenFold team found when trying to reproduce the AlphaFold 2 training. This performance is also certainly due to the limited size of the datasets used to train it. I would not be surprised, in fact, quite the opposite, if one of Isomorphic Lab’s key goals was to fine-tune, or even retrain, this model on their proprietary datasets. But more on that later…
The advances, outlined
There is one final major difference between AlphaFold 2 and AlphaFold 3: how it was validated.
AlphaFold 2 was released as a candidate to CASP14, a biennial community assessment where computational biologists try to predict the structure of several proteins whose structure has been determined experimentally — yet not publicly released. In AlphaFold 3, however, the authors have chosen not to use a similar gold standard validation, partially because for some of their categories (e.g. protein-ligand interaction prediction) there is not a good one. Instead, the authors have built test datasets from publicly available benchmarks, either already compiled, or built according to sensible rules from PDB data.
Using non-blinded data as test always entails a risk. Because we use the test data to evaluate the performance of the model, there is always the (well-intentioned) temptation to use this test data to select the best model. In other words, there is a bias towards models that perform well on the test data – something that can never occur with blind tests. I would encourage you to think about testing as predicting the stock market: would you rather trust someone who reports they predicted last week’s stocks very accurately, or someone who gives you the predictions for next week and lets you make your own assessment? For this reason, it is always advisable to assume that performance on reported test datasets is not necessarily as good as in a general case.
Protein-ligand prediction
The advance most people will be looking out for is protein-ligand prediction. Most drugs are small molecules, and most drug targets are proteins, so a model that can reasonably tell you where and how a molecule binds to a protein is bound to be (no pun intended) a powerful tool for drug discovery. But, how good is AlphaFold 3 exactly?
There are many benchmarks for protein-ligand complex prediction, but one that has become very popular recently is PoseBusters, proposed by OPIGlet Martin Buttenschoen. The benchmark contains a number of previously determined protein-ligand crystal structures, and evaluates the similarity between predicted and real poses. What makes the benchmark interesting is that it includes a variety of sanity checks (are the bond lengths sensible? does the molecule have steric clashes?) to make sure that the poses are physically sensible. The key metric is the percentage of generated, physically-meaningful poses, that are within 2 A of the real pose (an arbitrary threshold, but a well-established one).
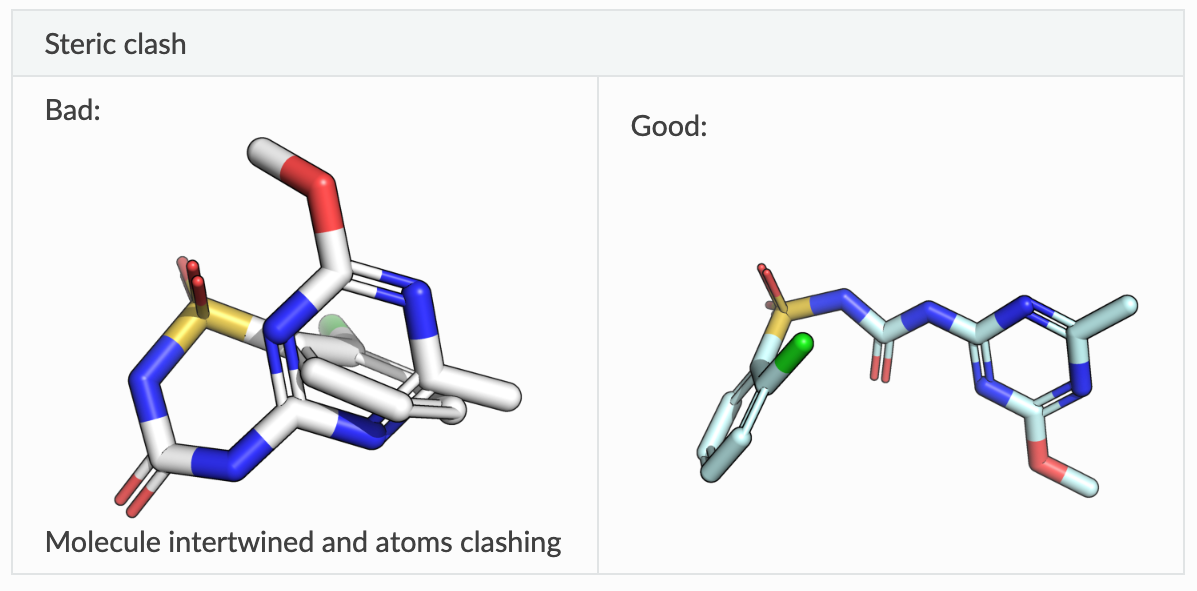
On the left, an example of a molecular pretzel, a common feature of some recent machine learning docking methods where the structure is entirely unphysical, even though quality scores (e.g. RMSD to crystal structure) suggest it is of good quality. On the right, the same molecule in a physically reasonable conformation. Image reproduced from the PoseBusters documentation.
In this benchmark, AlphaFold 3 achieves a performance of nearly 80%, which is significantly ahead of the second method, AutoDock Vina, standing at about 55%. While it is sensible to point out that AutoDock Vina is not the state-of-the-art (dates from 2015), it is one of the most well-established docking programs, and it is the top performer in the PoseBusters paper. Showing a significant improvement demonstrates both that AlphaFold 3 generates physically meaningful poses, and that, well, they are pretty similar to what one gets in the experiment.
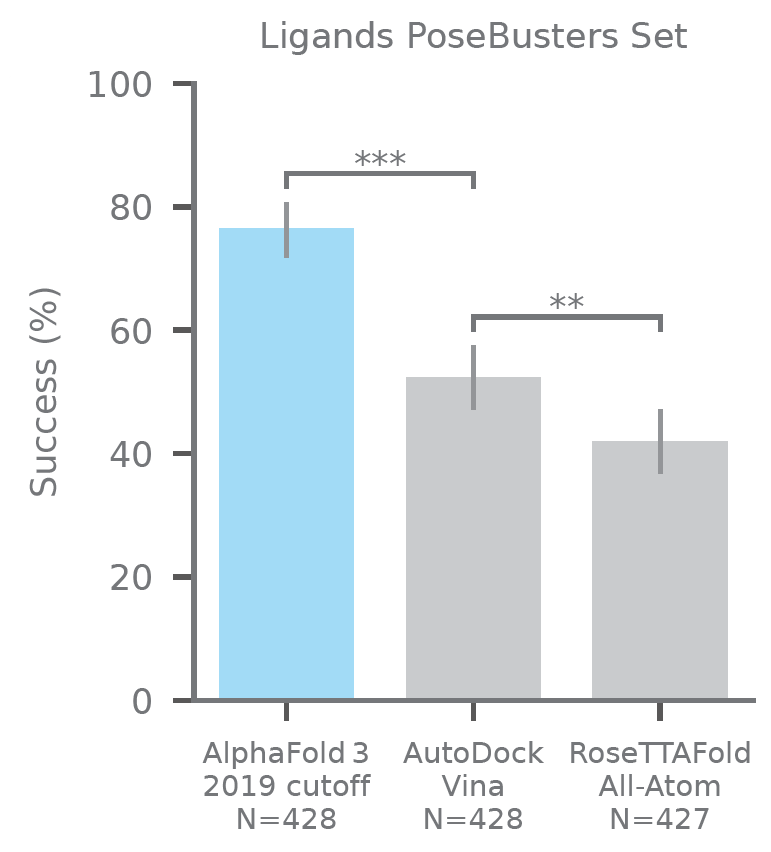
Performance of AlphaFold 3 on PoseBusters, a protein-ligand benchmark for machine learning docking. The y-axis indicates the percentage of successful models, which is percentage of models for which the RMSD of the pocket residues and the ligand is under 2 angstrom. Reproduced from Figure 1 in the ASAP article.
That said, all benchmarks in the protein-ligand interaction world are far from perfect. There have been many discussions about how they can be erroneous or incredibly biased. Most of the structures in the PDB will be proteins that are easy to crystallise and study, that have some therapeutic relevance, and that belong to a set of a few therapeutically relevant targets. Think about it: if you train a model on structures of kinase inhibitors, it will learn that anything that looks like ATP should go to the ATP binding site. For that reason, I find it shocking that the authors have not validated the model with crystal structures that have not been determined previously.
Another question is how limited these predictions are. The authors mention the case of an E3 ligase, which can adopt two different structures: an open state, in absence of the ligand; and a closed state, when bound to it. However, when running predictions of E3 ligases with and without the ligand, AlphaFold 3 always predicts the closed state. This is a somewhat disappointing outcome, as cofolding approaches – which have been discussed before, both in academic papers and in industry – have been posited to design drugs against targets with induced conformational changes, or even cryptic pockets that are only evident in one of the conformational forms of the protein.
The final, very practical incognita, is how long it takes to run the code. Molecular docking with a tool like AutoDock Vina takes of the order of a minute for a protein-ligand pair – this means if you want to evaluate a large library of potential drug-like molecules, like the 7 billion compound Enamine REAL library, you are probably looking at tens or hundreds of thousands of dollars in cloud CPU. Add now that you will need a pretty powerful GPU, and the numbers will skyrocket even further. This is not to say that AlphaFold 3 could not be incredibly useful for medicinal chemistry (even at multi-hour runtimes, it would be a terrific tool for lead optimization), but inference time will certainly determine its applicability.
Antibody-antigen predictions
Aside from small molecules, there is one specific class of proteins whose interactions with other proteins are very relevant for drug discovery: antibodies, the most common class of biotherapeutics. Antibodies are immune proteins that have uncanny specificity, and that neutralize pathogens or target specific molecules for destruction by the immune system. These properties have made them into extremely versatile pharmaceutical candidates, for example by finding a membrane receptor that is overexpressed in cancerous cell and targeting them for destruction by the immune system (trastuzumab, anyone?).
Antibody structure prediction was something that AlphaFold 2 struggled with. Most of the antibody sequence consists of a fixed template (or rather, one of a very limited set of templates), with a small variable region, which determines specificity. The variable region is generated in response to the presence of an antigen due to a combination of gene recombination and directed evolution. Because these processes do not leave out an evolutionary history, there is no benefit to using coevolution. This is the reason why it has been possible to develop deep learning models that outperform AlphaFold 2 for antibody structure prediction. For this same reason, AlphaFold 2 did not have particularly good performance at predicting the interaction between an antibody and its structure. In short: it cannot predict the epitope (the part of the target protein that is bound by the antibody) particularly well, and even when it can, the interaction of the side chains with the epitope does not resemble reality.
AlphaFold 3 seems to achieve a performance of antibody-antigen interactions that is roughly similar to the performance of AlphaFold 2 Multimer on general protein-protein interactions. The structures shown in the paper’s figures appear to be of high quality, and based on the reported interaction scores, it looks like there is an improvement in epitope identification. Howwever, a glaring omission is that there are no reports of the quality of side chain predictions in the model. This is quite important as knowing which specific interactions determine specificity will be quite important for protein engineering e.g. for producing variants that resemble a therapeutic candidate, but with improved solubility, stability or immunogenicity.
There is another interesting point and that has to do with the number of predictions needed. The authors report that “to obtain the highest accuracy, it may be necessary to generate a large number of predictions and rank them”. In one of the figures (see below), the authors authors show an enormous increase in performance, nearly double, when they chose the best (in terms of their predicted confidence) of 1,000 generations, rather than a single generation.
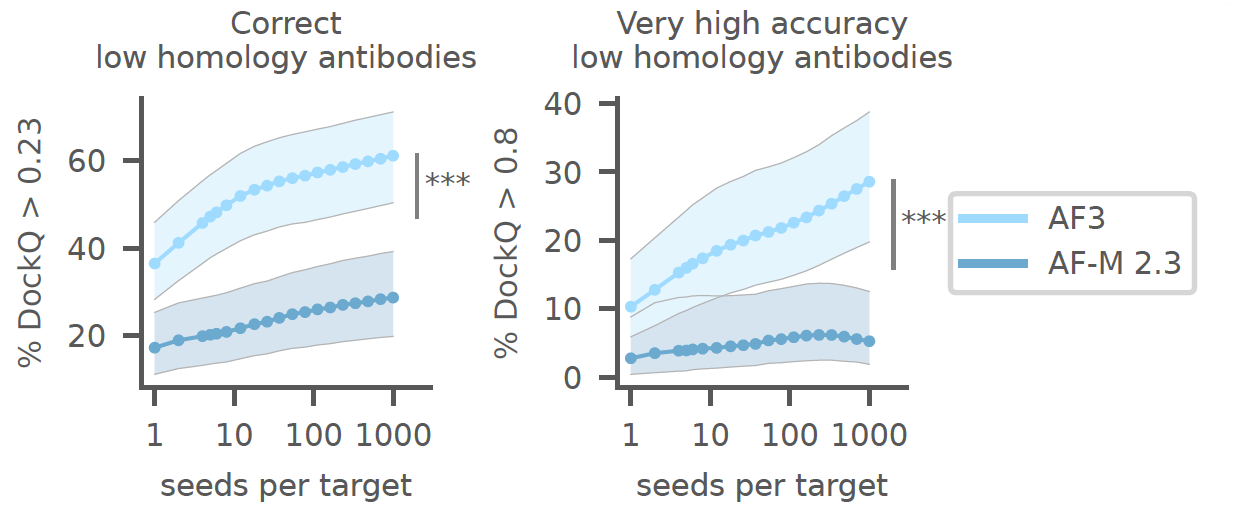
Sensitivity of AlphaFold 3 to the number of seeds in antibody-antigen prediction tasks. The y-axis indicates the percentage of successful models, which is defined as the LDDT for the interface. Reproduced from Figure 5 in the ASAP article.
The results seem quite promising. Antibodies are a hot topic in both industry and academia, and getting better at predicting how antibodies interact with their antigens is definitely a step forward in making better drugs and diagnostics. That said, there is quite a lot to clarify first. The good news is that the AlphaFold Server imposes no restrictions on proteins, so I am sure there are some groups already working on benchmarking AlphaFold 3 for antibody structure prediction tasks.
Protein-nucleic acid prediction
Another interesting question in biomolecular modelling is how exactly proteins interact with nucleic acids. There are manifold applications, but one of the key ones is to study how transcription factors interact with DNA. In short, transcription factors are proteins that bind to promoters, regions of the DNA sequence that precede genes and determine how much a given gene will be transcribed. These transcription factors in turn regulate themselves in complex ways: for example, a transcription factor can regulate how another transcription factor is produced, leading to an intricate regulatory network. There is a very significant clinical implication here, as many of the main proteins implicated in cancerous diseases are transcription factors (some of your favourite proteins: Myc, Rb, etc.)
The idea of using AlphaFold-like technology to predict protein-DNA interactions is not new, and the state-of-the-art was RoseTTAFold2NA, which was an extension of RoseTTAFold applied to protein-nucleic acid interactions. The authors validate AlphaFold 3 against RoseTTAFold2NA on a variety of PDB structures that contain protein-DNA and protein-RNA interactions, showing an excellent improvement. The authors also validate their model on 10 publicly available RNA crystal structures derived from CASP15. While AlphaFold 3 does not outperform the best method, Alchemy-RNA, it is remarkably close (see below), and it is promising that the only competitor to AlphaFold 3 is a method that relies on human inputs.
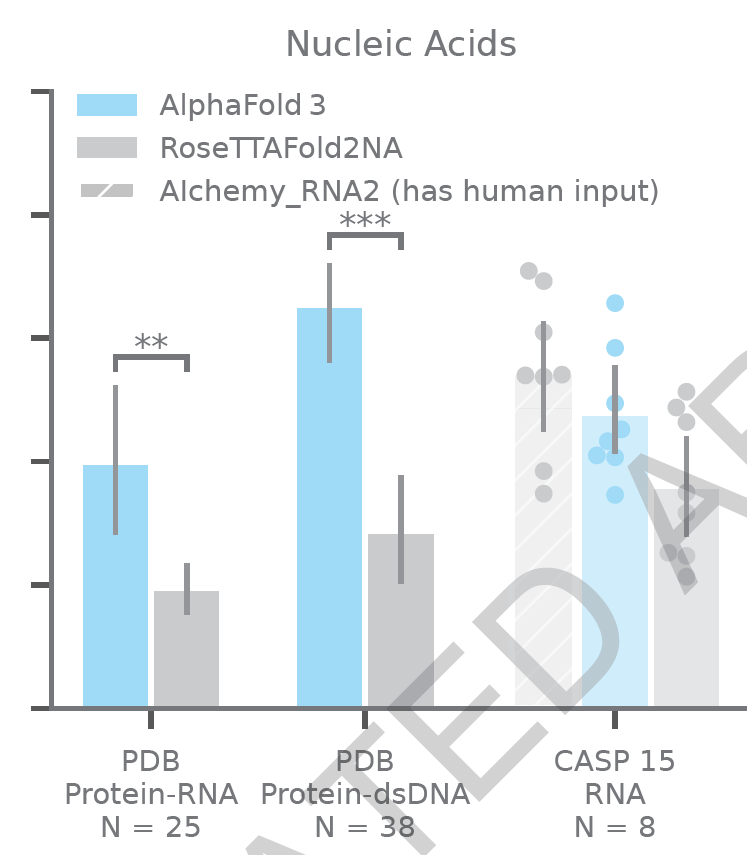
Performance of AlphaFold 3 on protein-nucleic acid benchmarks. The y-axis indicates the percentage of successful models, which is defined as the LDDT for the interface, except for the RNA case, where it is just the LDDT of the RNA monomer. Reproduced from Figure 1 in the ASAP article.
There is an interesting scientific point here. With AlphaFold 2, when you tried to predict the structure of a transcription factor, you would quite often get a lot of spaghetti, save for perhaps a clear structure corresponding to a conserved motif (e.g. a zinc finger). On the other hand, when predicting the structure of a transcription factor with DNA, the structure is better defined – in accordance with a central tenet of intrinsically disordered proteins that they adopt reasonable structures when in the presence of their binding partners.
Within my limitations as a non-nucleic acid expert, the structures look legit, quite what I would expect from protein-DNA structures. There is a good possibility that the transcription factor-protein structures result from the augmentations from JASPAR. All in all, it looks like a valuable tool for hypothesis generation.
Some other point: post-translational modifications
I am not going to discuss this last point in depth, but I want to at least mention that one of the final AlphaFold 3 selling points is its ability to predict non-canonical amino acids. Non-canonical amino acids are amino acid derivatives that typically arise from modifications after the protein is made, but can also result from artificial modifications. This ability to account for these unusual amino acids allows AlphaFold 3 to create more accurate models of protein structures, and also look at a new range of problems.
For example, you might have heard of one Hollywood star drug called Ozempic. Taking a few licenses, the mechanism of action of Ozempic might be simplified to a non-canonical amino acid mimicking the natural gut hormone GLP-1. This engineered amino acid can’t be broken down by enzymes as easily as the natural one, leading to a longer-lasting effect. This strategy is just one of the many in the playbook of peptide therapeutics, which is a growing area of biotherapeutics.
More broadly, post-translational modifications are crucial for understanding the biological behaviour of biomolecules. Antibodies, for example, are heavily glycosylated at specific positions, and this modification is crucial for both the stability and signal ability of the antibodies. Many proteins in the proteome are regulated by phosphorylation: adding a highly negatively charged group induces conformational changes. Acetylation, methylation, and ubiquitination are just a few other examples of these chemical tweaks that can dramatically alter a protein’s function. The ability of AlphaFold 3 to incorporate non-canonical amino acids will definitely contribute to better our understanding of biology, although it remains to establish how well exactly the predictions correlate with reality (for example, how well does AlphaFold 3 differentiate the phosphorylated and unphosphorylated versions of a protein).
The matter of the code
There is an issue with this whole story that has opened passions amongst the AI for biology community: the publication comes without code or weights. While the paper provides a wealth of information in the form of an extended supplementary information, pseudocode is definitely not the same as code. Even the AlphaFold Server, which is limited to twenty predictions a day, only allows a very reduced number of ligands. In other words: the science cannot be reproduced, the results cannot be verified, and the findings cannot be built upon to produce new scientific knowledge. This is a pretty appalling level of reporting for what could be one of the most exciting scientific breakthroughs this decade.
I want to make it clear that my complaint is not with Deepmind or Isomorphic Labs, which I think have done nothing wrong – within their duty to their shareholders, they have done more than enough to release valuable information. The duty to uphold scientific publishing befalls to the journal, which should deny publication if there is not sufficient detail to validate and reproduce the work. There is already an open letter to the Editors asking for explanations. There is no excuse – Roland Dunbrack, who was one of the reviewers, already talked on X about how he asked the journal to request the code, and received no response. I do not know what happened behind the scenes, or whether the code will be made available eventually. However, the thought that Nature enables an article to sidestep the comments of a reviewer seems sincerely worrying.
I want to remind you that Nature is one of the leading scientific journals, and has classically been one of the most important sources of scientific information. Watson and Crick published the structure of DNA there. So did the authors of many paradigm-shifting breakthroughs. Sure, it is impossible to be a top generalistic scientific journal and not be part of a controversy from time to time, but in general, if you read something on Nature you expect that it has been subjected to the highest standards of scientific peer review. At the very least, you expect a thorough check for potential issues like lack of reproducibility. Even in cases where the authors omit the training code, the weights, or even the data preprocessing pipeline (as in AlphaFold v1), there are possibilities to learn or reproduce the work within sensible means.
The precedent set by this paper is simply unacceptable. If you have spent any time reading techbio papers, you must have come across some crappy ones who make unreasonable claims and then do not release their code – likely, because they could not be reproduced. Don’t get me wrong, I am convinced that Deepmind/Isomorphic have done legitimately incredible work here. But, if it becomes commonplace that some can publish their work without being subject to the standards of peer review, how can we know what is true?
There are rumours in the scientific community that Deepmind has already approached some academic labs to make AlphaFold 3 available. There are also rumours that AlphaFold 3 will be made available for general academic usage within the next six months. What isn’t entirely clear is how exactly this will be made available, if as code or as a server, and there are significant differences there. While a server will enable lots of basic biology research, the code will be crucial if we expect incremental scientific progress on AI for biology.
If I had to bet, I would expect Alphabet to make AlphaFold 3 available as a server or an API, in the spirit of NVIDIA’s BioNeMo. This service will be free, or very cheap for academics, but it isn’t unthinkable that they also make AlphaFold 3 available for commercial use within some limitations. These limitations may range from some stark “no ligands” cut-off, to simply making the model in AlphaFold 3 available for everyone, while keeping their internal, more advanced models (both what they have been cooking since they finished this work, and the models that are trained in their internal datasets) for their drug discovery business.
Final discussion
My first impression after reading the AlphaFold 3 paper is that it was groundbreaking yet, somehow, less of a paradigm shift than the previous generation. When AlphaFold 2 came out, its capabilities were established beyond any reasonable doubt by its performance on CASP14, a blind test. The architecture provided many new innovations – as Mohammed AlQuraishi described it, “a beautifully designed learning machine, easily containing the equivalent of six or seven solid ML papers but somehow functioning as a single force of nature”. AlphaFold 3 has provided neither. No validations were provided beyond public benchmarks. And, while the architecture is brilliantly executed, we are missing some of the deeply permeating ideas that were brought about by AlphaFold 2.
A big reason behind this petering out of performance is likely to be the data. The datasets used in AlphaFold 2 and AlphaFold 3 differ by about 2 and half years of data collection (April 2018 for AF2, September 2021 for AF3), or roughly 30,000 new structures, a 20% dataset growth – probably much less once you remove structural redundancy. There has been a lot of discussion about how the sophistication in AlphaFold 2 was about finding the right inductive biases to understand a limited amount of data. The AlphaFold 3 paper is rich in clever ways to improve upon the previous architecture, but it is perhaps unsurprising that, with less data to start with, there is less additional information to distill.
I believe strongly that the next innovations in the field of biological AI will be brought upon, not by engineering neural networks, but by finding ways of generating massive amounts of data and putting them into the architectures. There is every reason to believe that this is what is happening at Isomorphic Labs (and many of their competitors) right now. While they may not be able to generate large amounts of structural data, they must be generating some additional information, just like they did with DNA. They may be exploiting their partnerships with big pharma to gain access to some of their structural databases. More likely, they will be crossing AlphaFold 3 predictions against massive DNA-encoded library screens to crack the problem of affinity.
I think we are about to witness a decade of extraordinary improvements in artificial intelligence and biology. I am convinced that we are about to see machine learning models significantly augment the capabilities of discovery scientists, partly by contributing to hypothesis generation, partly by replacing lengthy wet lab assays with high-quality predictions. It is not that AI is going to revolutionise biology overnight, I don’t think it will do that. But I am certain that AI will significantly lower the cost of drug discovery bets. And with more bets being made, some of them will pay off big.
I also think that a lot of these improvements will happen behind closed doors, in the insides of companies that can afford training the massive models, not to mention collecting the massive datasets. I anticipate that a lot of the developments in the next decade will be made in industry, rather than academia.
My undeniable techno-optimism should be tempered down by some reasonable constraints. As Derek Lowe would say AlphaFold 3 is great – but it doesn’t solve clinical trials yet. Most reports of AI drug discovery have been unsuccessful. The machine learning chain breaks by its weakest link: the relative lack of clinical data. We can collect large datasets of biophysics, and even preclinical studies, but someone has yet to find how to collect clinical data cheaply and at scale. Even in other areas beyond therapeutics (diagnostics, agritech, you name it), there are lots of barriers that lie in our lack of understanding of biology.
Yet, no matter the challenges, it certainly is an interesting time to be in techbio.