How large language models are shaping modern protein science
This blog post started as a collection of thoughts for a talk I gave for Pistoia Alliance, a non-profit organization encompassing hundreds of life sciences companies with the objective of increasing innovation, in June 2023.
You’ve probably heard this on your Twitter or LinkedIn feed, but I’m here to tell you again: large language models (LLMs) are a big deal. Even amidst the whirlwind of AI developments in recent months, these tools have positioned themselves at the heart of our workflow. Walk through any computational research lab and you’re sure to spot screens filled with ChatGPT tabs, writing code snippets, answering questions that are difficult to write in “Google form”, or even lending a hand with writing tasks. I admit it, this very paragraph might have gotten a little touch-up from ChatGPT itself.
While many understand the potential of LLMs to change the future of work, the impact of this technology in basic life sciences research has been much underappreciated. For one, biological LLMs are actually superhuman. Everyone speaks at least one language, and there are many that can produce quality text — but not even the most seasoned protein scientists could predict protein structure from sequence alone, or design a protein with a specific function. In fact, even the most advanced computational methods prior to biological LLMs were still far less successful.
In this blog post, I will provide a high-level tour of the field of protein language models (pLMs, though I abhor the acronym). My goal is to make this post approachable for everyone, but rather than spinning another jargon-heavy narrative, I aim to break down the technical aspects into the simplest terms possible. More specifically, I will be explaining how protein language models work, exploring their capabilities, and predicting how they’ll keep driving remarkable advancements in computational protein modelling. Hope this is a pleasant journey!
- How does a protein language model work?
- Generating protein sequences
- Protein language models and protein engineering
- Towards biological integration
- Thoughts and outlook
1. How does a protein language model work?
TL;DR. Protein language models are essentially identical to text LLMs, but they have been trained on databases of protein sequences instead of text. If you pop the hood of systems like ProtBERT, ESM, or ProtGPT2, you will find that the architecture has only minute differences with natural language processing systems like BERT or GPT.
So much for making it accessible and non-jargony, right? Okay, let’s go back to basics.
If you have ever tried to formally learn a second language, you might have come across exercises like the following:

This exercise, termed as the cloze task, serves as the foundation for LLMs’ learning mechanism. These models digest colossal volumes of text data (ChatGPT, for instance, reputedly ingested tens of terabytes of Internet-sourced text), which is then artificially corrupted to incorporate random gaps. The model is then confronted with the task of filling in these gaps. If the model fills them correctly, the neural network will be rewarded; otherwise it will receive a penalty and be nudged towards better future predictions. By repeating this process on a grand scale, with massive models, extensive computing power, and abundant text data, we arrive at systems that can accurately fill in the gaps, and even generate new text that abounds with the emergent properties we witness in ChatGPT et al.
If this sounds simple… well, it is. You can implement a functional GPT-like language model in a few hundred lines of code.
Right about now, you might be arching an eyebrow, thinking this seems too easy. I remember my own high-school English classes — filling in the blanks was really all about recalling the new words from the last unit. How does such a simple task capture complex aspects of language and grammar? Well, there’s quite a bit of linguistic finesse at play here. For instance, you won’t take long to rule out verbs, adverbs, or anything that isn’t a noun to fill the gaps:

Fine, you will say. Learning grammar is one thing — we have had grammar checkers for decades. It is quite another story to learn the subtleties and common sense in language. Yet, if you think about scaling the cloze task to vast amounts of text data, it quickly becomes obvious that some completions are far more likely and reasonable than others:

Dig a bit deeper, and you’ll convince yourself that the simple task of completing a sentence (as done by autoregressive models like GPT or the LLaMA series) or plugging in missing words here and there (like masked language models such as BERT) can unlock vast linguistic insights. We’re not just talking syntax and grammar, but a profound understanding of language. The key insight is that, even though text itself follows a straightforward one-dimensional pattern (one word after the next), the training strategy primes the model to discover the complex, long-range interactions between words that give rise to the emergent properties of language.
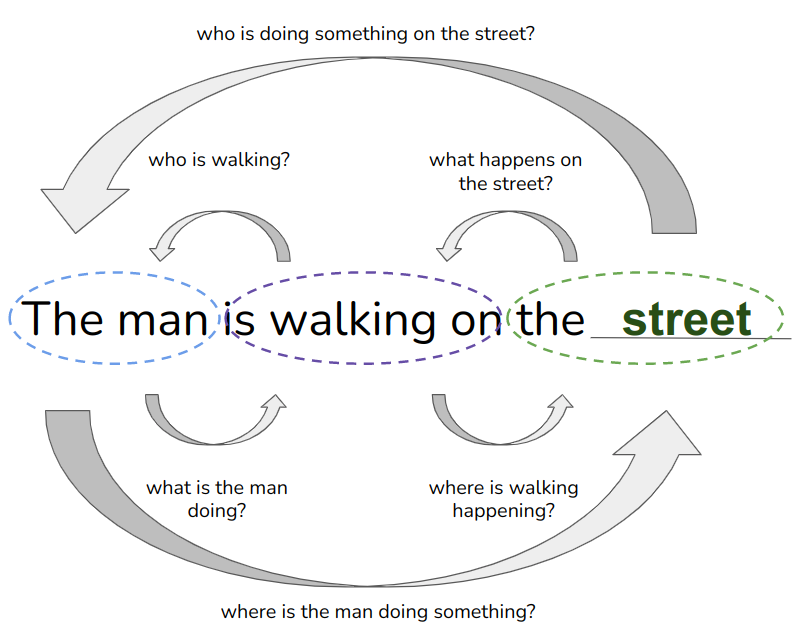
If you are with me, the analogy to proteins is almost immediate. Whereas we typically represent proteins as one-dimensional sequences of amino acids, proteins adopt complex three-dimensional structures that give rise to long-range interactions between parts of the sequence. Proteins also have common word-like arrangements of letters (secondary structure motifs) and syntactic units (domains). And, like language, small changes often lead to unchanged biological meaning, but can sometimes have dramatic effects.
Can we then use the same strategies to train language models that learn the complex biology at play in protein sequences? The answer is a resounding yes.
2. Generating protein sequences
Synthetic biologists often find themselves echoing Richard Feynman’s mantra: “What I cannot create, I do not understand”. If that adage is true, then we have a lot to learn about proteins. The complex fitness landscape of protein sequences, coupled with the vastness of the search space, makes it challenging to design sequences that behave according to our expectations or that even mimic natural proteins. It is no wonder that generating new protein sequences has been one of the first tasks handed to protein language models.
Protein sequence generation has mostly been done with autoregressive models in the fashion of the GPT series. There are many interesting papers in this area, and I would definitely not be able to do justice to all of them. In the interest of brevity, I will focus on two papers that I found particularly groundbreaking: the ProtGPT2 model by Noelia Ferruz and co-workers, and the ProGen model by Salesforce Research.
Both models use the same basic transformer decoder architecture, inspired in GPT2, and are trained on a subset of the UniProt Archive, on the task of predicting the next amino acid in the sequence. The models are fairly large, with 36 layers (though, due to differences in the embedding size, ProGen has about ~30% more parameters). After pretraining for what must have been a long time, the authors generated a large number of artificial sequences and tested their properties.
In the case of ProtGPT2, one of the first papers to harness LLMs for protein sequence generation, the authors zeroed in on the model’s potential to generate sequences that, while novel, were still biologically plausible. They examined sequence similarities to UniRef, observing that while there was homology, the sequences were novel and not just memorised. These artificial proteins were also predicted to be mostly globular, with well-defined secondary structures and levels of predicted disorder comparable to natural proteins. They also used very relatively unreliable (but still better than nothing) stability predictors like Rosetta scores or force field energies, to conclude that the generated proteins seem reasonably stable.
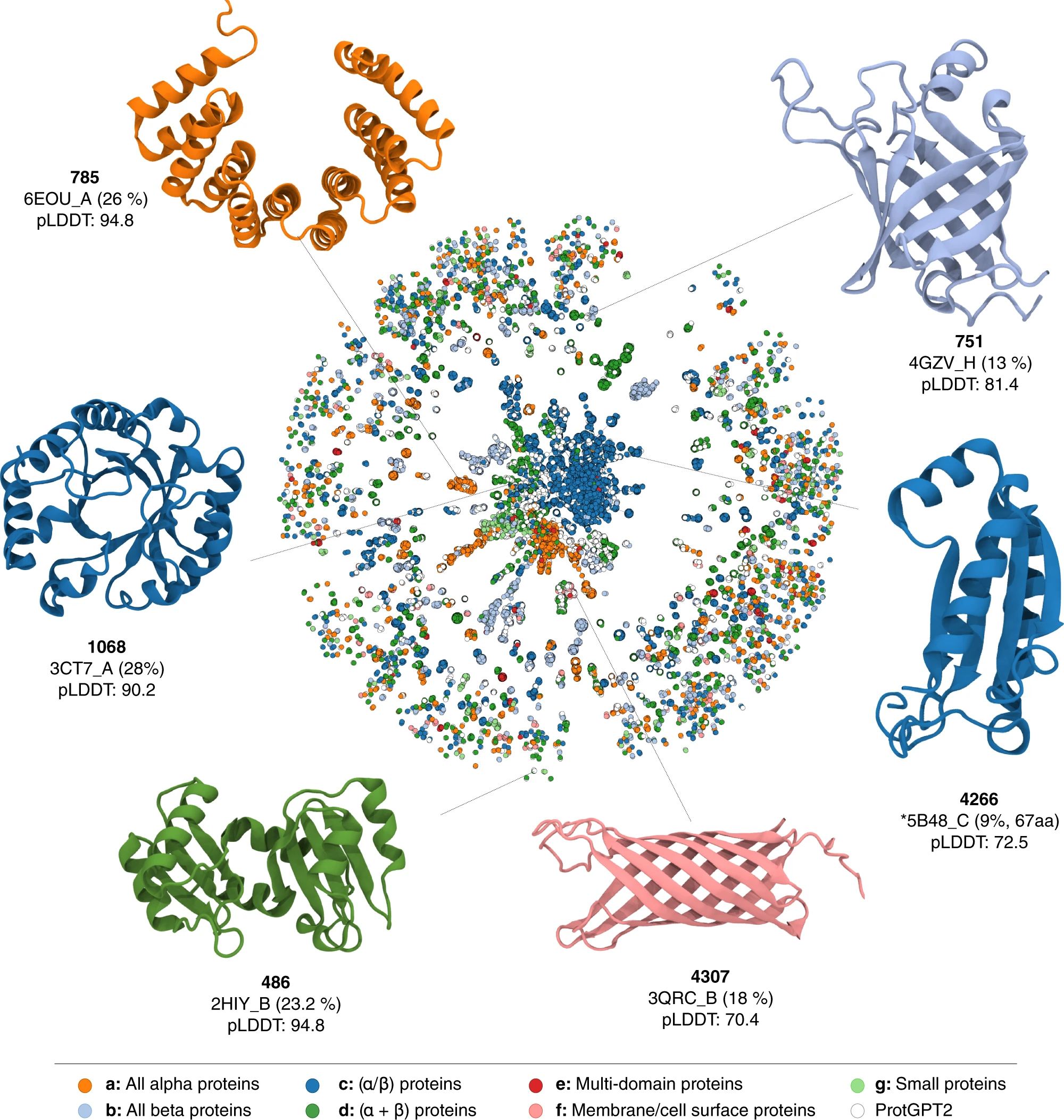
Network representation of protein space, showing that ProtGPT2 can generate proteins in all major classes, but also completely novel proteins.
Designing sequences is interesting — but we are not interested in just any sequence, only on protein sequences that have a specific function. This need has motivated methods like ProGen, that introduce a conditional tag for protein generation. At training time, the model is fed not only sequences to predict the next token of, but also some labels that represent taxonomy and functional properties (essentially Gene Ontology terms) of the protein. The objective is, of course, to provide these control tags to the model at inference time to generate sequences with the desired properties.
The ProGen authors use their model to design several protein sequences with enzymatic activities, and embark in an authentic tour de force on experimental validation of machine learning methods. For example, the authors finetune their model on a lysozyme dataset, then express a selection of generated sequences and demonstrate experimentally that ~75% of them exhibit the expected enzymatic activity. They even went all the way to obtain a crystal structure for one of the designed lysozymes that exhibited the strongest activity, and show that the structure is similar to the original lysozyme (albeit with a few changes on the substrate binding cleft).
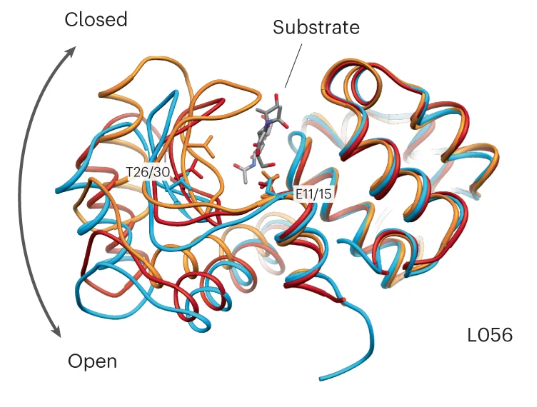
The crystal structure of the artificial lysozyme L056 (sky blue) determined by the Salesforce Research team, as well as crystal structure of T4 lysozymes in their open (dark red) and closed (orange) conformations.
Whatever you think of the results, it is clear that protein language models are promising tools to design novel proteins. But, what else can they do?
3. Protein language models and protein engineering
While generating protein sequences is certainly an exciting endeavour, there is far more potential in language models than just generating sequences in some protein family.
A common undertaking in language modelling is the creation of learned representations. The idea is to transform discrete entities – in this case, pieces of text – into continuous, high-dimensional vectors. There are various incentives to do this. For example, if the representation is devised in a way that the continuous space mirrors the structure of the data, we can anticipate that data with similar properties, like texts covering the same topic, will be in close proximity within this embedding space – enabling efficient search and retrieval. More crucially, discrete data, such as sequences or graphs, is not differentiable, a quality essential for the vast majority of (or, at least, the most effective) machine learning techniques. Hence, representations provide a method to incorporate this otherwise incompatible data into machine learning systems
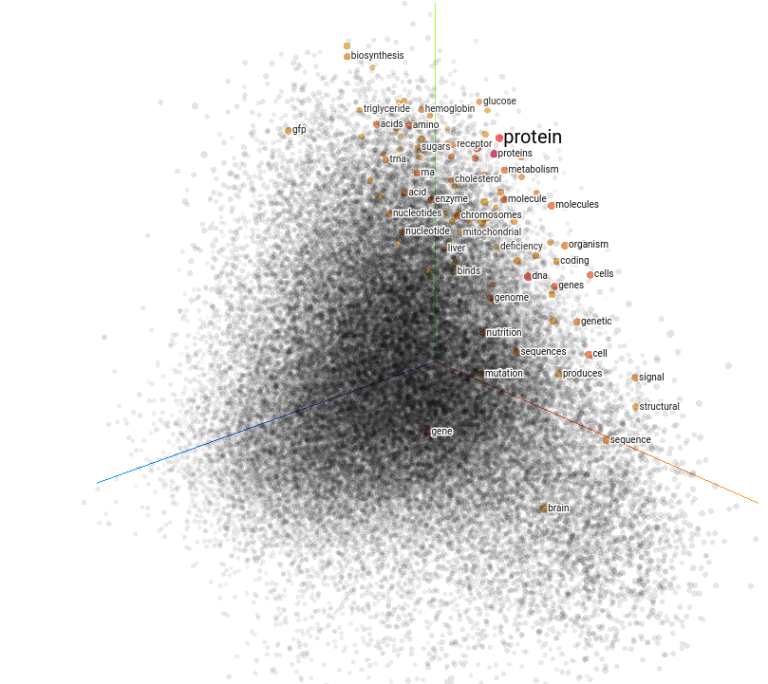
Visualization of the Word2Vec embedding, one of the classical representations in natural language processing. I selected the word “protein” and marked a number of words that show high similarity. This image was created using the TensorFlow Embedding Projector.
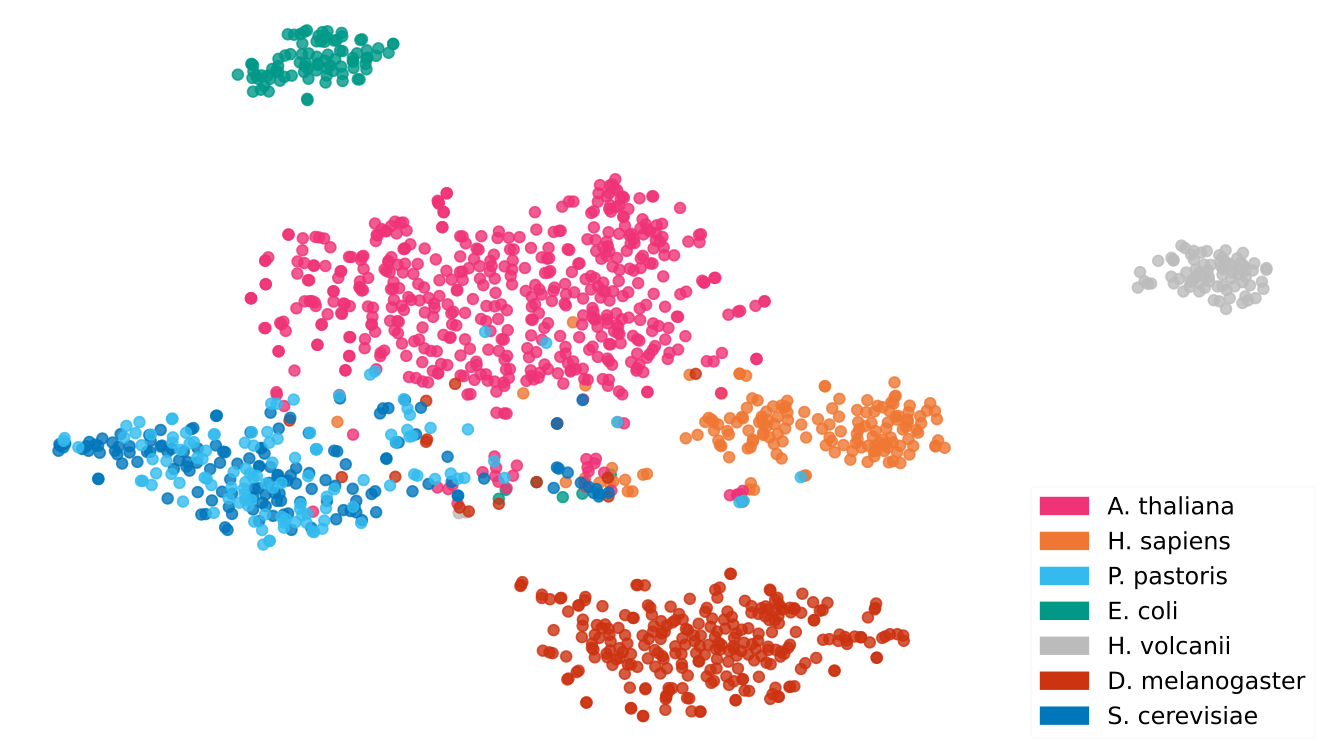
You know how the story goes — it just turns out that LLMs are also great at producing learned representations of proteins. In fact, this is one of the most common applications of LLMs in protein science. As of the time this article was published, representations from language models are central to the state-of-the-art tools for predicting protein function (see this and this), subcellular localisation, solubility, binding sites, signal peptides, some post-translational modifications, and intrinsic disorder, among others.
Let’s not overlook the fact that learned representations offer more than just a channel to integrate proteins into machine learning models. The real question is how we ought to represent a protein. Traditional property prediction papers have explored a wide range of tactics: one-hot encoding of protein sequences, position-specific scoring matrices, predicted physicochemical properties such as surface accessibility or isoelectric point, with varying levels of success. Yet, growing empirical evidence suggests that learned representations are decidedly superior. The intuition here is that these models, having been pretrained on a large unlabelled corpus, have the capability to extract the defining attributes of protein sequences that truly represent them, even though these may not be immediately perceptible to us humans. Think of these models as ‘distilling’ the quintessence of a protein into an abstract high-dimensional vector.
There are two primary strategies to tackle a property prediction task using learned representations. The first strategy hinges on using the language model solely as a featurizer. The vector, with no modifications, is fed directly into the model. The premise here is that the pretraining process will have equipped the network with the capacity to identify significant features in the protein sequence. The second approach, known as fine-tuning, necessitates training the “featurizer protein language model” in tandem with the machine learning predictor. This method is based on the idea that the language model will, consequently, evolve to generate representations that are not merely generic for protein-related tasks but tailor-made for the specific task at hand.
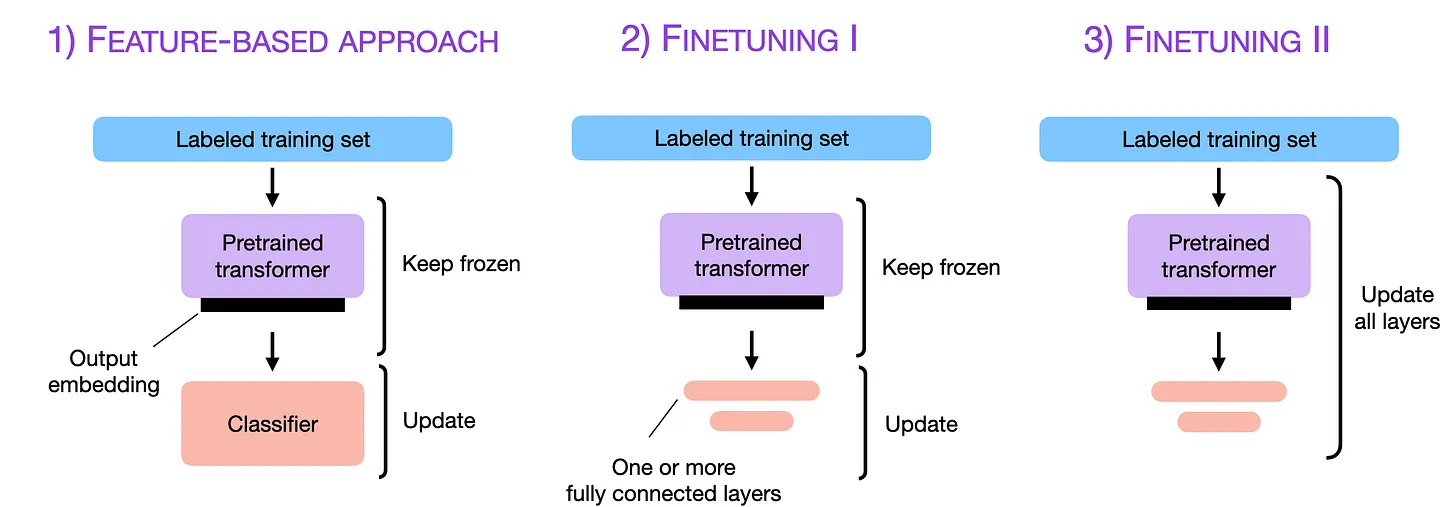
Image originally by Sebastian Raschka, whose excellent post on finetuning LLMs I can only recommend.
Both methods come with their respective pros and cons, some of which are fairly self-evident. Fine-tuning, which requires training the whole network, is decidedly more resource-intensive. One key challenge is the sheer size of these large language models — models like ESM2-48, boasting 15 billion parameters, won’t fit on a standard consumer-grade card; a data center-grade card such as an A100 would be required. Even then, one would be grappling with a really small batch size — meaning lengthy fine-tuning times. Of course, there are some tricks to accommodate the models in memory, like quantizing weights, gradient checkpointing, and many others. However, these solutions invariably come with their own set of trade-offs — extended training times, compromised precision, and the like. As usual, there are rarely free lunches.
Despite the fact that protein language representations are still far from perfect, and that research is being carried out to try to improve their representational power, it is undeniable that they are becoming an indispensable tool for protein engineering.
4. Towards biological integration
Thus far, we’ve explored applications that essentially take established techniques from language modelling and retrofit them for protein science. Protein property prediction, for instance, bears a resemblance to sentiment analysis, while protein generation mirrors text generation almost exactly. However, some clever applications have repurposed protein language models to tackle entirely different types of problems. We will look at some of these in this section.
Protein structure prediction, one of the most captivating problems in biology, is a particularly fascinating area where protein language models show promise (if you want to read more about protein structure prediction and machine learning, see my lecture notes). You may well know that AlphaFold 2 made a huge breakthrough in protein structure prediction. However, it’s important to remember that it relies on multiple sequence alignments (MSAs) to garner coevolutionary information. Constructing these MSAs can be challenging; they require significant computational resources, and in some cases (such as for artificial proteins or antibodies), they are simply not meaningful. Now, since the MSAs are used to extract information about long-range interactions in the protein sequence, and this is the very information that we expect protein language models to have learnt, some researchers have thought of using pLMs to replace MSAs in structure prediction algorithms.
The pioneering work in this domain can be attributed to Mohammed AlQuraishi’s lab. However, the most noteworthy examples today are perhaps the OmegaFold model from the American startup Helixon, and the ESMfold model by Meta AI. These models function by replacing the Evoformer block in AlphaFold 2 (did I mention my lecture notes on the subject?) with some amalgamation of a language model’s output. In the case of ESMfold, the AlphaFold 2 architecture is modified to take in the sequence embedding and the attention maps, and is then fine-tuned for protein structure prediction. While the results are quite promising, and have enabled predictions of very many metagenomic properties, these models have shown suboptimal performance with orphan proteins. This has led some authors to suggest the language models are just a standin for memorising multiple sequences alignments.
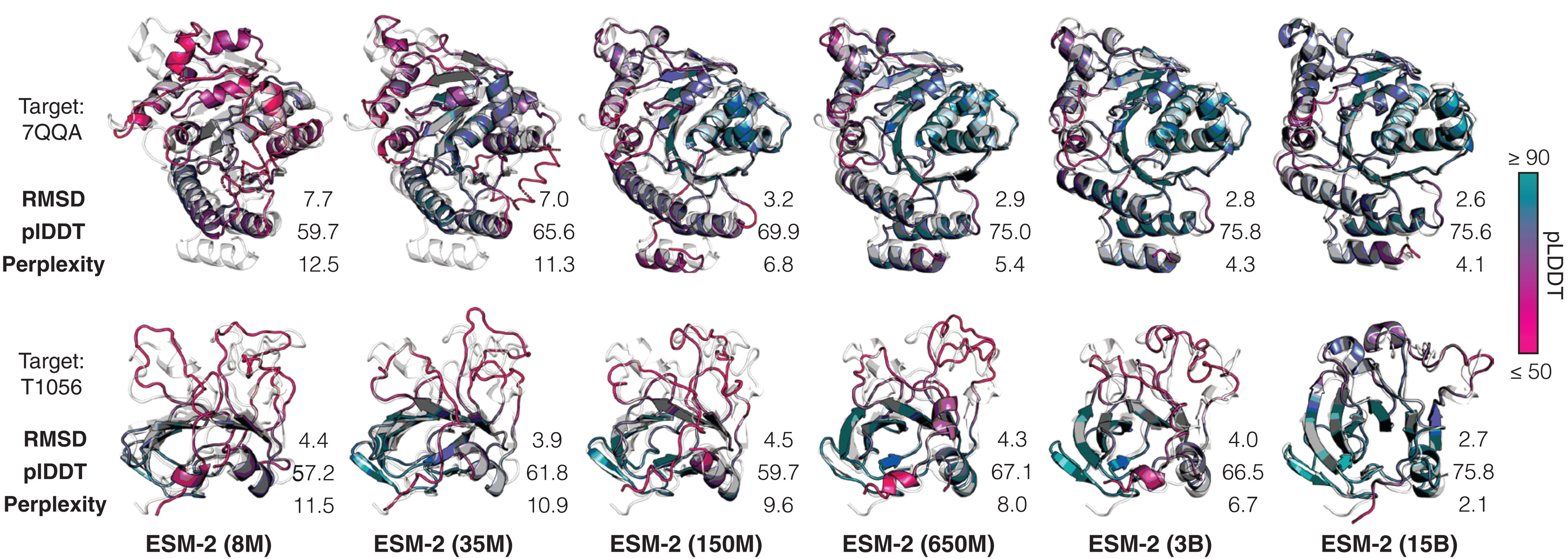
Panel from a figure in the ESMfold paper showing that the predictive accuracy of their pipeline improves as the language model's understanding of protein sequences improves.
Another compelling application of protein language models lies in fitness prediction: predicting whether a mutation at a particular site in a protein will bring about a significant functional change. This is a good question because — as often happens in science — we can collect fitness data using high-throughput experiments like deep mutational scanning. What is the general idea? Start with a large segment of DNA and copy it using an error-prone polymerase that introduces random mutations, and then transfect it into cells, generating a population of individuals with different versions of that protein. If the protein under study plays a vital role in cellular function (otherwise, what’s the point of studying it, right?), any mutations that impair its function will consequently negatively impact the cell’s survival rate. By allowing the cells to proliferate, or exposing them to certain conditions (say, an antibiotic, is the protein is expected to provide resistance), different cell populations will thrive or perish based on the protein’s functional efficiency.
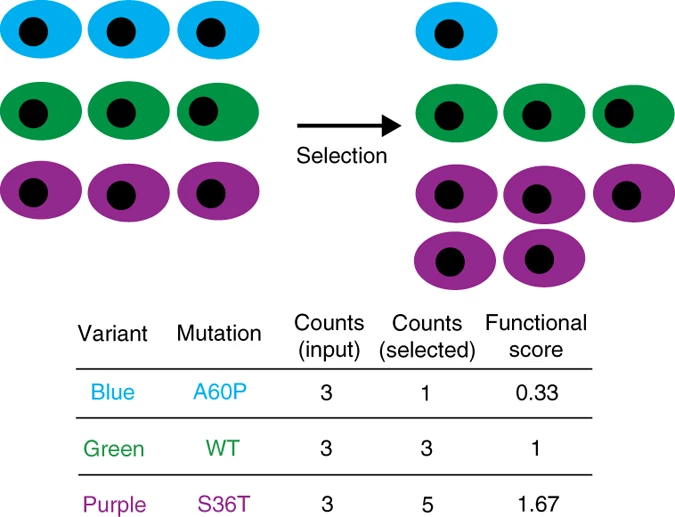
The selection condition enriches the sample in protein variants that thrive under this constraint, allowing to quantify fitness scores. Image reproduced from the Nature Methods paper by Fowler et al.
We define “fitness” as the relative proportion of cells with a specific protein in the final sample compared to the original sample. Since language models can predict the probability of a specific amino acid at a particular position, they can be leveraged to forecast the fitness of a mutation. Although there are slight technical variations between different methods, they generally hinge on the ratio between the wild-type amino acid’s probability and the mutant amino acid’s probability. Predictions can also be made directly from the pretrained language model (zero-shot), or using a model fine-tuned on experimental data.
Fitness predictions derived from language models display impressive correlation with results from deep mutational scans. Now, of course, while deep mutational scans serve as a convenient benchmark, the real value of these techniques extends far beyond — from predicting mutations that could potentially cause disease, to those that could help viruses elude immune responses.
To finish up, I’d like to brieflydiscuss the expansion of protein language models beyond proteins themselves, venturing into the realms of higher-order systems biology. A topic particularly dear to me is understanding how DNA encodes a protein. As you might know, there are 20 amino acids, but 61 potential combinations of DNA nucleotides (codons) that can encode these building blocks. This means that some amino acids can be represented by up to 5 different codons — there is significant degeneracy. The choice of codons is not random, and it’s been well-established that it’s intricately connected to the protein’s function, greatly influencing both protein folding and expression.
Last year, we published a paper demonstrating that training a large language model on codon sequences, instead of amino acid sequences, resulted in improved performance across a variety of tasks. Regardless of the tasks we examined, the codon-trained model consistently outperformed all amino-acid-based models of similar parameter count (keep in mind, we’re an academic group — our 80M parameter model can’t compete with Meta AI’s 15B model!). However, we managed to show that our model significantly outperformed all others in several tasks. We’re currently exploring how to utilize this model for novel engineering tasks and for gaining insights into regulatory systems biology.
These are just some of the examples of how language models are becoming crucial tools in computational biology. More than just a clever method for featurizing proteins or generating analogous sequences, protein language models are rapidly evolving into integral components of sophisticated algorithmic architectures that can tackle some of the most challenging problems in biology. And there is much, much more to come.
5. Thoughts and some future outlook
Protein language models (pLMs) are one of the most exciting developments in artificial intelligence applied to biology. Unlike text-based LLMs, which amaze us with their human-like eloquence, pLMs are mastering tasks that already surpass even the abilities of top-tier human experts. I am convinced that the potential of pLMs is enormous, and that we are only scratching the surface of what they can do. And there is much, much more to come. Better architectures that incorporate biological insight, better, more informative data, and the side benefits of the recent push in text LLM research will all contribute to the development of better pLMs.
In this picture, data is poised to be a key determinant. As model size begins to approximate or even exceed the size of standard datasets (for instance, the 15B parameter ESM2 model surpasses the UniRef50 dataset on which it was trained!), the generation of more high-quality data becomes crucial. One company that I am excited about (no shares or conflicts of interest, rest assured) is Basecamp Research. Their team embarks on expeditions to remote locales (such as Antarctica and the Amazon), gathering samples and sequencing as many proteins as possible. Similar approaches, such as automated pipelines that can test the function and properties of thousands of proteins, will also be crucial to extract the maximum amount of power from the language models.
Yet undoubtedly the next significant leap in pLMs will come from the development of more sophisticated architectures that incorporate biological insights. Note that the success of language models to date largely hinges on scale: huge datasets, huge models, and huge computation time. Also huge financial investments. However, this trajectory of success is likely flattening out. While sufficiently deep pockets might one day train a 100B protein language model (for comparison, GPT-3 has 175B parameters), the pace of gathering more protein sequence data is unlikely to keep up. The only viable path forward is to embed biological insights directly into the models, thereby boosting their capacity to learn from data more efficiently. Clever design, incorporating both biological and AI insights, holds the key for the future of pLMs.
Despite the promise, there are many open questions. What is the best way to integrate protein language models into more complex architectures? How do we make our data more informative? How do we make sure that the models are learning physics alongside biological relations? How do we make the models more interpretable? How do we make training more efficient? How do we find the best finetuning recipes? How do we… you get the gist, there is quite a lot to do in the field of protein language models, and I suspect we will only see more and more exciting advances in the coming few years.